|
|
Acer 30-32. 08038 BARCELONA |
|
 |
|
|
|
Reactivos de Derivatización PIERCE |
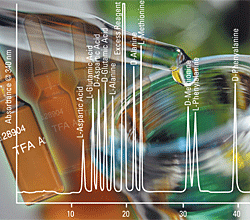  .gif) |
|
DERIVATIZACIÓN EN EL ANÁLISIS DE AMINOÁCIDOS |
El desarrollo del análisis
de aminoácidos empezó en 1820 cuando Braconnot aisló la glicina de un
hidrolizado de gelatina1. En 1848 Mulder demostró que la
glicina contenía nitrógeno2 y en 1883 Kjeldhal propuso un
método para la determinación precisa de nitrógeno contenido en una
muestra de proteínas o aminoácidos3. 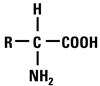 A
partir de 1910 la mayoría de los aminoácidos han sido aislados y se han
descrito sus estructuras. A medida que aumentaba el número de
aminoácidos fue posible agruparlos en base a características químicas
comunes. En esa época se descubrió que todos los aminoácidos tienen la
misma fórmula general y difieren solamente por la estructura química de
sus cadenas laterales. A
partir de 1910 la mayoría de los aminoácidos han sido aislados y se han
descrito sus estructuras. A medida que aumentaba el número de
aminoácidos fue posible agruparlos en base a características químicas
comunes. En esa época se descubrió que todos los aminoácidos tienen la
misma fórmula general y difieren solamente por la estructura química de
sus cadenas laterales. El análisis de aminoácidos efectuados a principios de 1900 eran muy laboriosos, a veces de semanas de duración. A medida que se descubría el contenido en aminoácidos de un número de proteínas, la información exacta no siempre se podía obtener dada la instrumentación disponible en la época4. La introducción de la cromatografía abrió nuevas vías para el análisis de los aminoácidos. Martin y Synge aportaron el primer ejemplo mediante cromatografía de partición, que separaba los acetil derivados de ciertos aminoácidos5. En este método se establece un equilibrio entre dos fases líquidas. Se mezcla gel de sílice en una solución de agua y un indicador. La mezcla se empaca en una columna como fase estacionaria. Los aminoácidos acetilados se disuelven en un disolvente creando la fase móvil. Los ácidos se introducen en la misma columna y los acetil derivados migran a diferentes velocidades. La separación se hace visible por el cambio de las bandas de color del indicador. Aunque este método separaba con éxito los los ácidos mono-carboxílicos y mono-amino no resultaba práctico para otros tipos de aminoácidos. Más tarde Martin et al. usaron filtros de papel como alternativa a la gel de sílice, desarrollando un método de cromatografía sobre papel que todavía se utiliza hoy en día. Los aminoácidos se disuelven en butanol y se dejan permear el el papel de filtro durante un determinado período de tiempo. El papel se seca y se trata con una solución diluida de ninhidrina (2,2-dihidroxi-1,3-idandiona). Las marcas coloreadas se miden y se comparan con los valores establecidos para esas condiciones experimentales. Estas separaciones eran sólo semi-cuantitativas, mientras que la cromatografía en columna tenía potencial para desarrollar métodos cuantitativos, pero la separación era imperfecta. La introducción de la cromatografía de intercambio iónico resolvió esos problemas, permitiendo la separación en columna de los aminoácidos sin derivatización previa 6,7. Las mejores separaciones se obtuvieron con resinas polisulfónicas (como Dowex50)8. Las modificaciones de los procedimientos han mejorado las separaciones de aminoácidos. Las características de la resina, el tamaño y temperatura de la columna, el pH de los tampones y la fuerza iónica han ido siendo modificadas para mejorar la resolución de mezclas de aminoácidos y obtener separaciones específicas. La cuantificación se ha mejorado enormemente mediante reacciones post-columna con nihidrina y con o-ftalaldehído (OPA) para mejorar la sensibilidad analítica. |
|||
| Desarrollos en el Análisis de Aminoácidos | |||
|
Las mejoras en el análisis
de aminoácidos se han basado en el sistema analítico y en la
instrumentación. Se han desarrollado procesos analíticos que, en función
del pH del tampón o de la fuerza iónica, permiten desplazar los
aminoácidos en bandas discretas. Los sistemas tamponados son compatibles
con el análisis de los aminoácidos (en una o dos columnas) presentes en
hidrolizados de proteínas o fluidos fisiológicos y se controlan por el
contra-ión usado (Na o Li) y el gradiente de cambio de tampón
introducido en la resina9-15. El tamponante más usado, el
citrato resulta adecuado para soluciones por debajo de pH 716.
Desafortunadamente para trabajo a alta sensibilidad el ácido cítrico
contribuye significativamente a la contaminación de los aminoácidos, por
lo que para obtener análisis consistentes es esencial usar reactivos de
alta pureza para la preparación de tampones. Existen otras alternativas a la separación de aminoácidos por intercambio iónico: puesto que este tipo de análisis es una de las herramientas básicas en la química de las proteínas, se requieren métodos más rápidos y sensibles para la separación y cuantificación17. Dos de los métodos más usados actualmente se basan en derivatización pre-columna y cromatografía de Fase Reversa: la formación de derivados dansil18-19 o OPA20-24. Ambos métodos ofrecen mayor sensibilidad y análisis más cortos que las técnicas post-columna. Otros métodos incluyen derivatización de aminoácidos con fenilisotiocianato (PITC) y la separación y cuantificación de los derivados feniltiocarbonilo resultantes vía HPLC. Estos derivados son lo suficientemente estables como para no requerir técnicas de derivatización en línea. |
|||
| Preparación de las Muestras | |||
La extracción y
purificación de las proteínas juegan un papel importante en la
determinación de aminoácidos. Estos métodos se basan en una o más de sus
propiedades físicas (p. ej. solubilidad, tamaño molecular, carga,
polaridad e interacciones específicas covalentes o no covalentes. Las
técnicas más usadas en la separación de péptidos y proteínas incluyen:
La tabla siguiente muestra una lista más detallada de métodos para el fraccionamiento de mezclas de péptidos25. |
|||
| Técnica | Propiedades Explotadas de las Moléculas de Péptidos | ||
| Centrifugación | Solubilidad | ||
| Cromatografía de Exclusión por Tamaño | Tamaño | ||
| Cromatografía de Intercambio Iónico | Carga, y en parte polaridad | ||
| Electroforesis en Papel | Carga y Tamaño | ||
| Cromatografía en Papel | Polaridad | ||
| Electroforesis de Capa Fina | Carga y Tamaño | ||
| Cromatografía de Capa Fina | Polaridad | ||
| Electroforesis de Gel en Poliacrilamida | Carga y Tamaño | ||
| HPLC | Polaridad | ||
| Cromatografía de Gases | Volatilidad de los derivados | ||
| Extracción Selectiva | Polaridad, y en parte interacciones específicas | ||
| Cromatografía de Afinidad | Interacciones específicas | ||
| Cromatografía covalente o de enlace irreversible | Formación de enlaces disulfuro; reactividad de homoserina lactona | ||
| Hidrólisis | |||
|
La mayoría de las muestra
de proteínas requieren algún tipo de tratamiento químico antes de que
los aminoácidos que las componen sean adecuados para el análisis. Las
proteínas y los péptidos han de hidrolizarse a aminoácidos libres y en
general se usan ácidos, usualmente HCl, para este fin. Un procedimiento de hidrólisis simplificado se basa en el reflujo de la proteína con un exceso de HCl, y la eliminación del exceso bajo vacío20. La proteína liofilizada se suspende en Ácido Clorhídrico 6N de ebullición constante y se introduce en el tupo de hidrólisis. La muestra se congela sumergiendo el tubo en hielo seco y acetona. Antes de ser cerrado, el tubo se trata con vacío para evitar la formación de ácido cístico, sulfóxido de metionina y clorotirosina27. Este procedimiento minimiza la descomposición de la S-carboximetilcisteína reducida y analiza las proteínas S-carboximetiladas. La hidrólisis se hace a 110ºC (con un control estricto de la temperatura) durante 20-70h según el método de Moore y Stein28. Tras hidrólisis, el HCl residual se elimina en rotavapor. El residuo se disuelve en agua y llevado a pH adecuado en función de la columna de análisis28. |
|||
| Catálogos | |||
|
|||
| Referencias | |||
|
|||
Copyright© 2006-2014 CromLab S.L. Prohibida la reproducción total o parcial sin consentimiento escrito del propietario.